4. Probe selection options¶
The probes used to measure microarray expression levels in the AHBA data are
often redundant; that is, there are frequently several probes indexing the same
gene. Since the output of the abagen.get_expression_data() workflow is a
region by gene dataframe, at some point we need to transition from indexing
probe expression levels to indexing gene expression levels. Effectively, this
means we need to select from or condense the redundant probes for each gene;
however, there are a number of ways to do that.
Currently, abagen supports eight options for this probe to gene conversion.
All the options have been used at various points throughout the published
record, so while there is no “right” choice we do encourage using the default
option (differential stability) due to
recent work by Arnatkevičiūte et al., 2019 showing that it provides the highest
fidelity to RNA sequencing data.
Available methods for probe_selection fall into two primary families:
We describe all the methods within these families here. Methods can be
implemented by passing the probe_selection argument to the
abagen.get_expression_data() function. For a selection of references to
published works that have used these different methods please see the
documentation of abagen.probes_.collapse_probes().
4.1. Selecting a representative probe¶
The first group of methods aim to select a single probe from each redundant group. This involves generating some sort of selection criteria and masking the original probe by sample expression matrix to extract only the chosen probe, which will be used to represent the associated gene’s microarray expression values:

The only difference between methods in this group is the criteria used to select which probe to retain. The descriptions below explain how the mask in the above diagram is generated for each available option; in each diagram the red outline on the generated vector indicates which entry will be used to mask the original matrix. The extraction procedure (i.e., applying the mask to the original probe by sample expression matrix) is identical for all these methods.
4.1.1. Max intensity¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='max_intensity')
Selects the probe with the highest average expression across all samples (where samples are concatenated across donors).

4.1.2. Max variance¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='max_variance')
Selects the probe with the highest variance in expression across all samples (where samples are concatenated across donors).

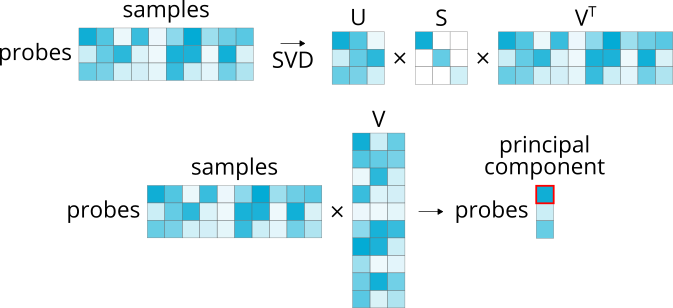
4.1.3. Principal component loading¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='pc_loading')
Selects the probe with the highest loading on the first principal component derived from the probe microarray expression across all samples (where samples are concatenated across donors).
4.1.4. Correlation¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='corr_intensity')
>>> abagen.get_expression_data(atlas['image'], probe_selection='corr_variance')
When there are more than two probes indexing the same gene, selects the probe with the highest average correlation to other probes across all samples (where samples are concatenated across donors).

When there are exactly two probes the correlation procedure cannot be used, and
so you can fall back to either the Max intensity
(corr_intensity) or the Max variance
(corr_variance) criteria.
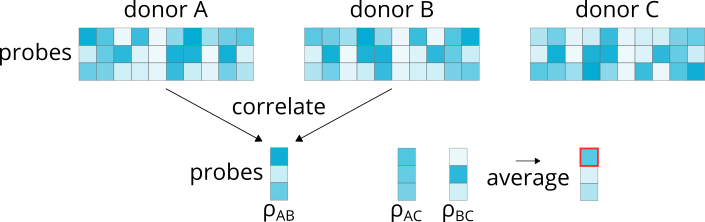
4.1.5. Differential stability¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='diff_stability')
Computes the Spearman correlation of microarray expression values for each probe across brain regions for every pair of donors. Correlations are averaged and the probe with the highest correlation is retained.
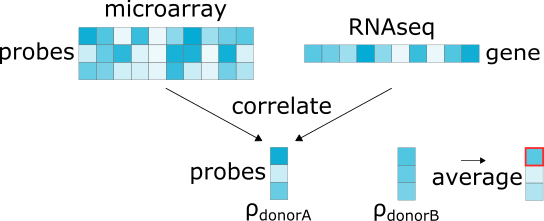
4.1.6. RNAseq¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='rnaseq')
Computes the Spearman correlation of microarray expression values for each probe across brain regions with RNAseq data for the corresponding gene. As only two donors have RNAseq data (donors #9861 and 10021), this method only computes the correlations for these two donors. Correlations are averaged across the two donors and the probe with the highest correlation for each gene is retained.
4.2. Collapsing across probes¶
In contrast to selecting a single representative probe for each gene and discarding the others, we can instead opt to use all available probes and collapse them into a unified representation of the associated gene:
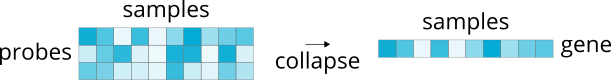
Currently only one method supports this operation.
4.2.1. Average¶
>>> abagen.get_expression_data(atlas['image'], probe_selection='average')
Takes the average expression values for all probes indexing the same gene.

Providing 'mean' instead of 'average' will return identical results.
5. Donor aggregation in probe selection¶
Unless otherwise specified in the description of that method, probe selection is performed using data aggregated across samples from all donors. However, this may not be desired: the probe that most reliably indexes a gene in one donor may differ from the probe that does so in another donor.
To allow for this possibility, we describe three options for modifying how
probe selection is performed across donors in detail below. These methods can
be implemented by passing the donor_probes argument to the
abagen.get_expression_data() function.
5.1. Aggregate selection across donors¶
>>> abagen.get_expression_data(atlas['image'], donor_probes='aggregate')
The default option, this will aggregate tissue samples from all donors and
apply the chosen probe_selection method to this single probe x sample
matrix. The probe chosen to represent each gene will be identical across all
donors.
5.2. Independent selection for donors¶
>>> abagen.get_expression_data(atlas['image'], donor_probes='independent')
Performs the chosen probe_selection method independently for each donor.
The probe chosen to represent each gene may be different across donors.
Note: this option cannot be used when the specified probe_selection is one
of: ‘diff_stability’, ‘rnaseq’, or ‘average’.
5.3. Most common selection across donors¶
>>> abagen.get_expression_data(atlas['image'], donor_probes='common')
Performs the chosen probe_selection method independently for each donor and
then uses the most commonly-selected probe to represent each gene. The probe
chosen to represent each gene will be identical across all donors.
Note: this option cannot be used when the specified probe_selection is one
of: ‘diff_stability’, ‘rnaseq’, or ‘average’.